RELX Opens Bioscience Lab to Enhance E-Cigarette Research
RELX Technology has started operations at its newly established e-cigarette bioscience laboratory to conduct systematic research on the effects of e-cigarettes through in vivo and in vitro tests, as well as conduct pre-clinical safety assessments.
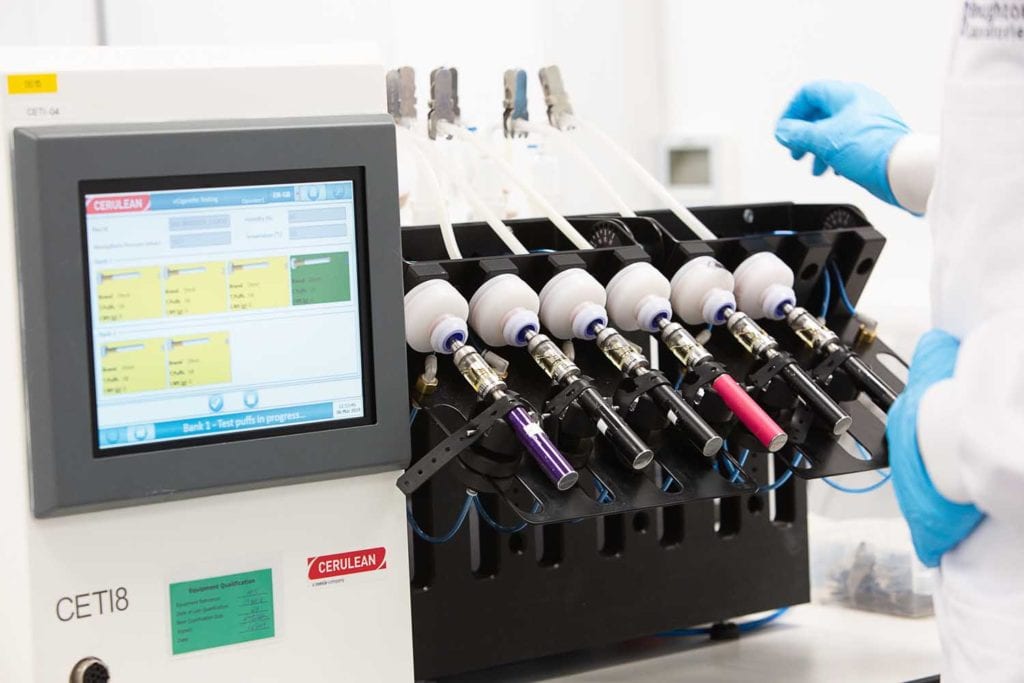
Located in Shenzhen, China, the bioscience laboratory is currently conducting research on the impact of RELX products on animal cardiovascular, respiratory, and nervous systems, to better carry out a comprehensive impact evaluation of vapor products.
“Science is the foundation of trust. As the industry leader, we have the responsibility to expand the borders of e-cigarette science and explore the unknown,” said Kate Wang, founder and CEO of RELX, in a statement.
At the company’s recent lab open day, RELX also announced its plan to establish a “1+4” scientific research approach—i.e. anchored by platform development, followed by toxicological assessment, clinical assessment, perception behavior study and long-term assessment.
“E-cigarettes are sometimes viewed with suspicion because we have incomplete knowledge,” said Yilong Wen, RELX Co-founder and head of science, research and development and supply chain, “The RELX bioscience lab’s mission is to explore the unknown. We want to collect evidence through a scientific approach and strive to prove the potential for e-cigarettes to be less harmful, and in doing so, provide users the option to choose an alternative.”
To ensure the reliability and quality of its products, RELX established a chemical and physical laboratory in 2018. The laboratory is certified by the internationally recognized China National Accreditation Service for Conformity Assessment.
RELX is currently conducting research projects on different topics with six universities including the Sun Yat-sen University and Shenzhen Institutes of Advanced Technology, the Chinese Academy of Sciences, two hospitals and nine scientific research institutions.