Why the World Cannot Afford America’s Regulatory Model
By Dr. J. Preston Campbell, Cancer Researcher, Harm Reduction Scientist
Heavy lifetime smoking kills approximately half of long-term users. That is not a projection or a model output — it is one of the most replicated findings in twentieth-century epidemiology, confirmed across decades and continents (WHO, 2023). In the United States alone, tobacco-attributable disease generates an estimated $310 billion in annual healthcare costs, roughly $6,500 per smoker per year (Xu et al., 2015; ASH, 2021). Globally, the burden exceeds $1.4 trillion annually (WHO, 2023). We know what is causing the damage. We have products that demonstrably reduce it. The question worth asking — the one this analysis is organized around — is why those products cost 167 times more per life saved to bring to market in the United States than in the European Union.
The Health Economics Punchline
Nicotine pouches are, by most objective measures, one of the less scientifically interesting harm-
reduction products ever developed. No combustion. No inhalation. No tobacco leaf. Pharmaceutical-grade nicotine salt in a cellulose pouch placed between lip and gum for thirty minutes. Peer-reviewed toxicant studies document 95–99% lower exposure to carcinogens compared to cigarettes (Mallock et al., 2019; Snusforumet, 2021). The first Cochrane systematic review, published October 2025, found no serious adverse events across all trials and confirmed consistently reduced toxicant biomarkers in every switching cohort studied (Hartmann-Boyce et al., 2025). The product’s risk profile is not controversial. What is controversial, apparently, is the cost of allowing it to be sold.
The standard health-economic metric for evaluating interventions is cost per quality-adjusted life year (QALY) gained — a single number that combines how long and how well someone lives. The U.S. cost-effectiveness threshold is $50,000–$100,000 per QALY. Published ICER estimates for comparable nicotine delivery products run $7,500/QALY for NRT sampling programs and $11,454/QALY for e-cigarettes used as cessation aids (Maciosek et al., 2022; Masiero et al., 2025). Based on the observed 5.2% switch rate in national survey data and the documented 95% risk reduction from switching, nicotine pouches are estimated to fall in the $3,000–$15,000/QALY range — well inside the cost-effectiveness frontier by any standard (Delnevo et al., 2024; Goniewicz et al., 2025).
At that switch rate, applying regulatory entry costs to the lives saved annually from harm reduction produces Figure 1.
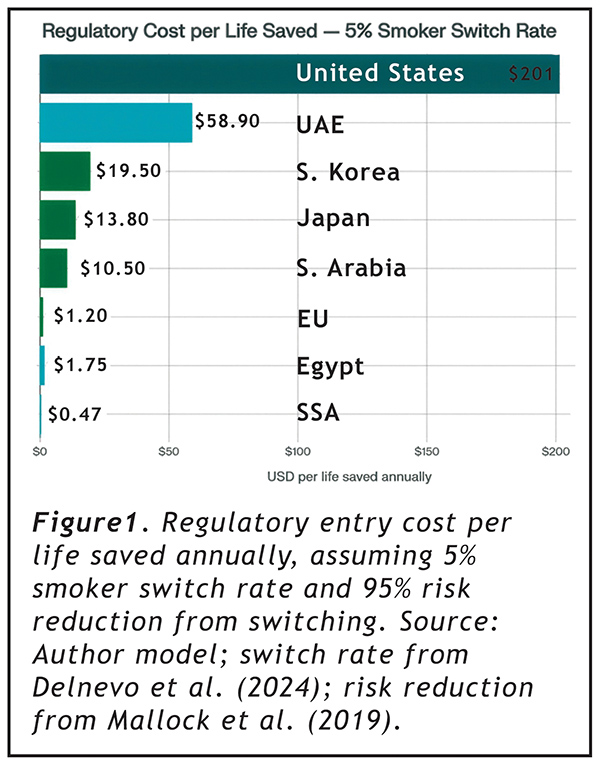
The U.S. costs $201 per statistical life saved per year. The EU costs $1.20. Sub-Saharan Africa costs $0.47. These numbers do not argue that FDA should wave products through without review. They argue something more specific: that a regulatory framework costing 167 times more per life saved than its closest comparator is not optimizing for public health outcomes. It is optimizing for something else.
Why: The Entry Cost Structure
The reason is straightforward once the numbers are placed side by side.
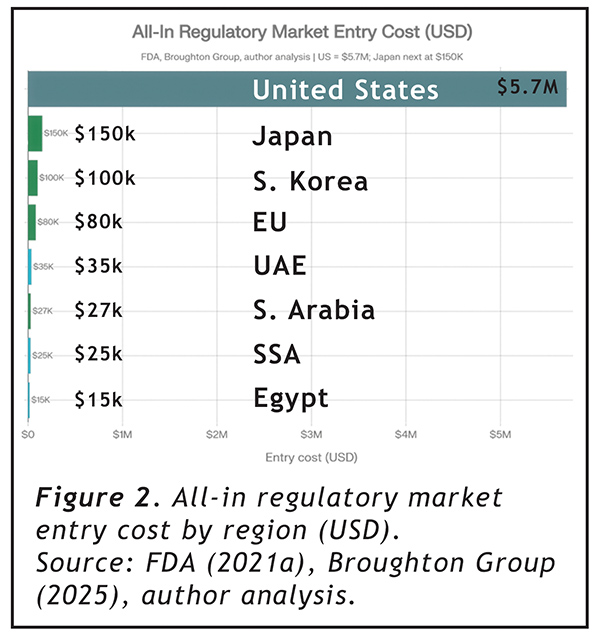
In the United States, bringing a single nicotine pouch product line to market requires a Premarket Tobacco Application. FDA’s own published cost estimate of $117,000–$466,563 per application (FDA, 2021a) has not aged well. Industry experience places the all-in cost at $4.2–$7.2 million per product family, once pharmacokinetic trials ($1M+), 12-month stability studies ($250K+), consumer perception research, addiction liability assessments, and regulatory consulting are properly accounted for (Broughton Group, 2025; JJCC Group, 2026). ZYN’s January 2025 marketing authorization — the first for this product class — took years and the resources of a Fortune 500 company (FDA, 2025a).
Elsewhere, the numbers describe a different regulatory universe. EU notification under the Tobacco Products Directive: $30,000–$80,000 per market. UAE ECAS registration: $35,000. Saudi Arabia: $27,000. Egypt — an open market of 14.5 million smokers — $15,000 (Broughton Group, 2025; SGS, 2025; author analysis). A company could simultaneously enter the EU’s 110 million-smoker market, Japan, South Korea, UAE, Saudi Arabia, Egypt, and sub-Saharan Africa for less than one U.S. PMTA submission.
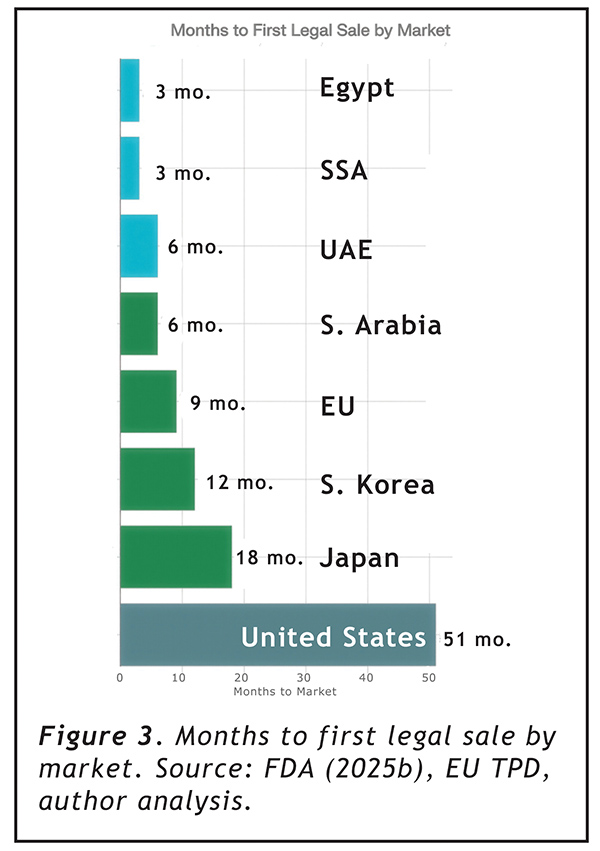
Why It Is Worse Than It Looks: The Time Factor
Entry cost alone understates the problem. The PMTA requires a minimum of 15 months of mandatory studies before submission, followed by an FDA review period averaging over three years to date (FDA, 2025b; Tobacco Law Blog, 2025). From the date a company decides to enter the U.S. market, the minimum realistic timeline to first legal sale is 51 months — over four years. The EU takes 9 months. The UAE and Saudi Arabia: 6 months. Egypt and sub-Saharan Africa: 3 months (author analysis).
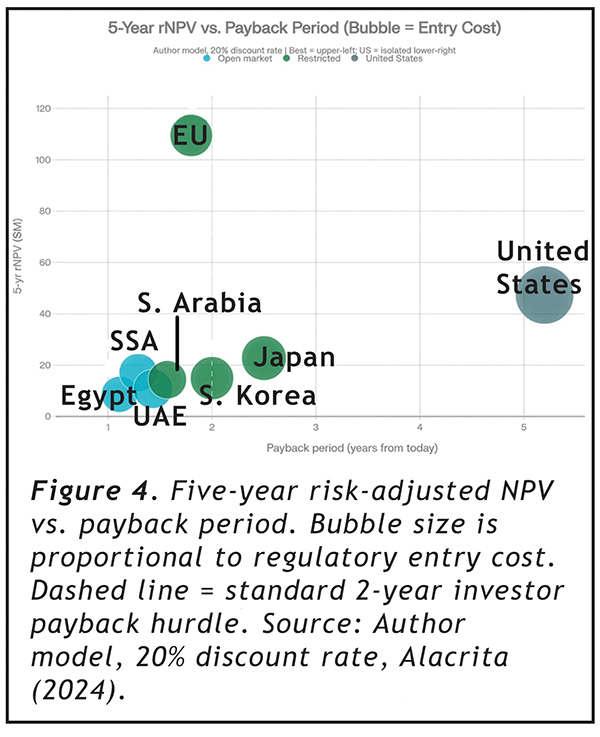
When those timelines are applied to financial models — discounting future revenue at the 20% rate typical for early-stage consumer products and adjusting for denial risk (estimated at 35% for new U.S. entrants; FDLI, 2023) — the picture becomes harder to rationalize from any direction.
The EU, despite having roughly half the U.S. annual revenue potential, generates a 5-year risk-adjusted NPV of $109.5 million compared to the U.S. at $47.3 million — because it begins generating cash 42 months earlier at one-tenth the cost. The U.S. payback period from today to regulatory cost recovery is 5.2 years, compared to 1.8 years for the EU and 1.2 years for Egypt. The U.S. is the only market in this analysis that exceeds the standard 2-year payback threshold that most venture and growth-equity investors apply to consumer product categories (Alacrita, 2024). During the 48-month excess waiting period, a company forfeits approximately $270 million in gross margin that would otherwise accrue in markets where the same product is already legally sold.
The 5-year ROI multiple on regulatory spend, risk-adjusted: 55× for the U.S., compared to 3,135× for the EU and 1,465× for sub-Saharan Africa. The PMTA does not just cost more — it structurally consumes the financial return from market participation, leaving only companies with institutional balance sheets able to absorb it.
The Cessation Paradox FDA Built
Here is where it gets genuinely strange.
FDA’s own authorization of ZYN stated explicitly that the products “have the potential to provide a benefit to adults who smoke cigarettes and would like to switch to a lower-risk alternative” (FDA, 2025a). The PMTA submissions included cessation and switching behavior data. FDA reviewed them. FDA relied on them to issue authorization.
And then FDA’s post-authorization communications carefully noted that the products are “not FDA approved” and that “there is no safe tobacco product” — language that systematically prevents companies from communicating the public health rationale that justified the authorization (FDA, 2025a).
Under the Tobacco Control Act, tobacco products cannot make cessation claims. If a company wants to say anything about reduced risk, it must file a separate Modified Risk Tobacco Product application — another process, more years, more millions, with public comment periods, advisory committee referrals, and post-market surveillance requirements (FDA, 2025c). As of 2024, FDA had received 48 MRTP applications since 2010 and authorized exactly 16 products (LDI Penn, 2025).
The logic chain deserves a slow read: FDA required switching studies as a PMTA condition. It reviewed them. It relied on them to grant authorization. It then prohibited companies from telling consumers those studies existed. The MRTP pathway functions in practice as a second admission ticket for a claim FDA already accepted as scientifically valid during the PMTA review. This is not a minor regulatory inconvenience. It is the architecture of a system in which harm reduction is simultaneously required as scientific evidence and forbidden as commercial communication.
What Rational Regulation Would Look Like
FDA’s September 2025 nicotine pouch pilot program demonstrated that rigorous and efficient review are not mutually exclusive (Tobacco Law Blog, 2025). The on! PLUS authorization in December 2025 confirmed it. Real-time applicant communication, literature-based study requirements, and focused critical element review cut timelines dramatically without any evidence of compromised standards (FDA, 2026).
That pilot should be the floor, not the ceiling. A product class with documented 95–99% lower toxicant exposure, no combustion, no inhalation, no tobacco leaf, and no serious adverse events in any trial conducted to date (Hartmann-Boyce et al., 2025) does not require the evidentiary apparatus of a novel pharmaceutical. The rest of the world has largely reached this conclusion.
In the meantime, 47.7 million Americans who smoke continue using a product that kills approximately half of them — while regulators elsewhere issue notifications, complete registrations, and authorize sales. At $201 per life saved, the U.S. PMTA process is not the problem. The problem is that it is 167 times more expensive than the alternative, without any measurable difference in the public health outcomes it is designed to protect.
_____________________________________
J. Preston Campbell, PhD, is a cancer researcher, harm reduction scientist, and co-founder of multiple companies, currently focusing on ENSO, his consulting and formulation company. His work focuses on translating reduced-risk nicotine products from laboratory science to regulatory approval and commercial deployment.
